

백혈병 표적항암제, ‘글리벡’ 내성 원인 유전자 찾았다
기적의 표적항암제’라고 불리는 ‘글리벡(성분명: 이매티닙)’에 내성을 일으키는 새로운 유전자가 밝혀졌다. 이 항암제가 잘 듣지 않던 30% 환자들을 치료할 수 있는 길이 열려 크게 주목받고 있다.
글리벡은혈액암 세포에만 발현되는 특정 표적을 공격해 부작용을 줄이면서도 치료 효과는 크게 높인 표적항암제다. 이 약의 개발로 만성골수성백혈병 환자는 골수이식이 필요한 심각한 상황에서도 장기생존이나 완치가 가능하게 됐다. 우리나라에도 2001년부터 이 약이 도입됐으며, 환자들을 하루 한 번 이 약을 복용하면서 암을 이겨낼 수 있었다.
하지만 이 약도 반복 복용하면 내성이 생겨 약효가 떨어졌고, 내성이 생기면 백혈병 암세포가 무한히 증식해 1년 이내에 사망할 가능성도 있었다. 게다가 만성골수성백혈병 환자의 10%는 처음부터 글리벡 내성을 가져 치료가 어려우며, 20%는 처음에는 잘 듣다가 내성이 생겼다.
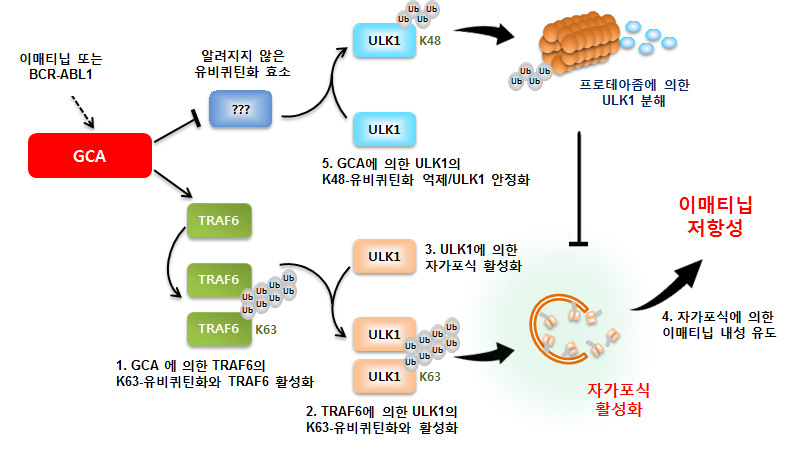
김홍태 생명과학부 교수팀은 같은 학부 명경재 특훈교수(IBS 유전체 항상성 연구단장), 김동욱 서울성모병원 혈액내과 교수팀, 이주용 충남대 분석과학기술대학원 교수팀과 공동으로 글리벡 내성을 조절하는 ‘지씨에이(GCA; Grancalcin)’ 유전자를 발견했다. 또 GCA 유전자가 TRAF6-ULK1 의존성 자가포식 작용을 활성화한다는 분자생물학적 기전을 찾아내 백혈병에 걸린 쥐를 모델로 이 내용을 규명했다. TRAF6 유전자와 ULK1 유전자는 서로 신호를 주고받으며 자가포식 작용을 활성화할 수 있는데 여기서 GCA 유전자가 기여한다는 것이다.
특히 GCA 단백질은 급성백혈병으로 진행하지 않은 환자에게서 발현이 증가하며 글리벡에 강한 내성을 보이는 데 관여하고 있었다. 원래 표적항암제 내성의 주요 원인으로는 BCR-ABL1 유전자의 점돌연변이가 알려져 있었다. 그런데 이런 기전은 급성백혈병으로 진행하지 않은 환자들에게는 아주 적게 발견됐다. 이는 곧 다른 내성 기전이 중요하게 작용할 가능성을 보여줬고, 연구진은 5년 동안 노력해 다른 주요한 내성 원인을 규명해냈다.
이번 연구에 따르면, GCA 단백질은 (1)TRAF6 단백질을 활성화시키며 (2)ULK1의 K63-연관 유비퀴틴화를 증가시켜 ULK1 단백질을 안정화시키는 동시에 활성화시켰다. 이런 작용은 결국 세포의 자가포식과정을 크게 증가시키므로, 표적항암제를 지속적으로 사용해도 백혈병 세포의 생존율을 높인다. 이런 분자생물학적 기전 때문에 글리벡 내성이 유지되는 것을 밝혀낸 것이다.
공동 교신저자로 참여한 김홍태 교수는 “이번 연구로 GCA 유전자가 지닌 저항성 유도에 관한 성질을 밝힐 수 있었다”며 “GCA 유전자가 만성 백혈병에 대한 치료제로서 기능할 수 있다는 가능성을 제시했다는데 연구의 의의가 있다”고 말했다.
김동욱 교수(가톨릭혈액병원장)은 “그동안 환자들이 글리벡 덕분에 백혈병은 중병도 아니라고 인식될 만큼 표적치료 효과가 높았으나, 환자 10명 중 3명은 약이 듣지 않았다”며 “이번 연구로 글리벡 내성이 어떻게 발생하는지 규명돼 새로운 진단법과 치료법 개발의 가능성을 크게 높였다”고 연구의 의미를 설명했다.
이번 연구는 의학과 세포 생물학 분야 세계 최고 학술지인 ‘오토파지 (Autophagy; IF=11.1)’ 3월 30일 자에 게재됐다. 연구 수행은 과학기술정보통신부·한국연구재단 바이오‧의료기술개발사업, 기초과학연구원(IBS), 한국백혈병은행, 대웅제약의 지원을 받아 이뤄졌다.
한편 공동 연구팀은 차세대시퀀싱과 마이크로어레이 방법으로 2017년 3월 만성백혈병이 급성백혈병으로 진행하며 차세대 표적항암제 타시그나 (성분명: 닐로티닙)에 내성을 획득하는 데 결정적인 역할을 하는 ‘코블 1(COBLL1)’ 단백질을 찾아 백혈병 분야의 세계적인 권위지 ‘루케미아 (Leukemia; IF=10.023)’에 발표한 바 있다.
Media link : [Unist] [biospectator] [Medical observer] [동아사이언스] [Medical Times] [헬스코리아뉴스] [etnews] [medworld] [HelloDD]